EZEN A KÉRDŐÍVEN várjuk az Angelman szindróma című kiadvánnyal kapcsolatos véleményeket.
Előre is köszönjük a visszajelzéseket!
Az Angelman szindróma előfordulási aránya 1/10.000, ill. 1/25.000 közé tehető.
Az Angelman szindróma egy genetikai eltérés által okozott idegrendszeri fejlődési rendelleneség, melyet értelmi fejlődési elmaradás, mozgás- és egyensúlyozási zavarok, a beszéd- és a nyelvfejlődés elmaradása és jellegzetes viselkedési sajátosságok jellemeznek.(1)
Harry Angelman 1965-ben számolt be először három gyermekről, akiknek hasonló széles mosolyú arckifejezésük, hasonló viselkedésük és tüneteik voltak. Az Angelman szindróma azóta ismert mint különálló kórkép.

Rajzoló gyermek portréja
Giovanni Francesco Caroto (1480-1555)
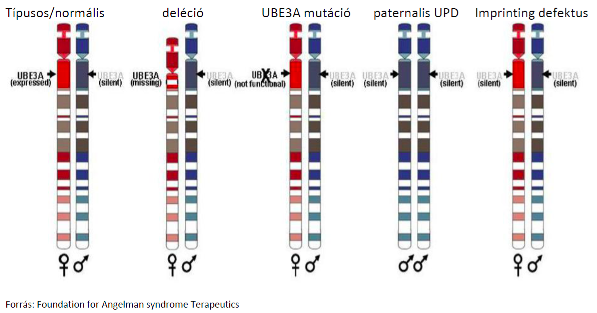
Angelman szindrómát okozó genetikai eltérések:
1. 70% -ban de novo anyai deléció a 15-ös kromoszóma hosszú karján (15q11.2-q13)
2. 25% -ban a fent említett területen található UBE3A gén mutációja okolható a betegségért
3. 2%-ban apai uniparenteralis disomia az érintett régióban (15q11.2-q13)
Az érintett génszakasznak mindkét kromoszómán az apai – inaktivált- génjei találhatók meg.
4. 3%-ban imprinting defektus (a meglévő genetikai állomány kifejeződésének szabályozási zavara –metilációs, hiszton acetilációs zavarok)(2)
Ezen kívűl elkülönítendő az Angelman-like szindróma (CDKL5, MECP2 gén mutációjával), illetve hasonló tünetekkel is járhat és elkülönítendő: pl. a Rett syndroma, ill. egyéb autizmussal járó rendellenességek, Christianson szindróma, MTHTF reduktáz szindróma, alfa-thalassaemia-retardációs szindróma.
A Prader-Willi szindrómáért felelős génszakaszok jelentős átfedéseket mutatnak az Angelman szindrómáért felelős génszakaszokkal (apai UDP Angelman, anyai UDP Prader-Willi szindrómát okoz), ennek megfelelően több közös pont is található a két betegségben (például értelmi érintettség, szőke vagy vörös/világos hajszín, hajlam a túlsúlyra). (1)
Angelman szindrómára jellemző klinikai tünetek:
– súlyos mozgás- és értelmi fejlődési elmaradás
– mozgáskoordinációs zavarok
– csökkent izomtónus
– epilepszia
– beszéd hiánya, vagy súlyos károsodása
Egy-két éves kortól külső tünetek:
– microcephalia (fejkörfogat növekedése meglassul, ezért a koponya kisebb lesz)
– az arc eltérései: arcközép alulfejlett, de szélesebb szájacska (macrostomia), előemelkedő áll (prognathia), fogak között emiatt kissé nagyobb távolság, előemelkedő nyelv
– világos haj, kék szem
– csökkent izomtónus, lazább végtagok
– babaszerű kéz- és csuklótartás
– egyensúly zavara, ülés-, járászavar
– szaggatott végtagmozgás
– torokköszörülés, nyelészavarok
– kancsalság, illetve szemészeti eltérések gyakrabban fordulnak elő
Viselkedési jellemzők:
– nagyon felhangolt, vidám viselkedés
– kirobbanó, nehezen értelmezhető nevetés
– kifejezett hyperaktivitás, mely serdülőkor végére jellemzően oldódik
– izgalmas dolgok megízlelése, szájba tömködése (mely az ízérzés kifejezettebb használatával magyarázható)
– impulzivitás és a víz kifejezett szeretete
– ha valami miatt izgatottak, akkor a másik személyes terét kevésbé tisztelik (pl. csipkedéssel hívják fel magukra a figyelmet)
– igen gyakran jelentkeznek kisgyermekkorban alvászavarok, feltöredezett alvási ritmus, felborult napi ritmus, mely későbbiekben javul
Értelmi fejlődés jellemzői:
– súlyos értelmi fejlődési elmaradás
– beszéd hiánya, vagy minimális formában jelbeszéd
– epilepsziás rohamok 80-96%-ban kisded korban jelennek meg (gyakori atípusos absence, generalizált tónusos-clonusos rohamok, atoniás rohamok, myoclonusos rohamok, nem ritkán komplex parciális rohamok, illetve a diagnózist megelőzően lázas görcsrohamok) (3)
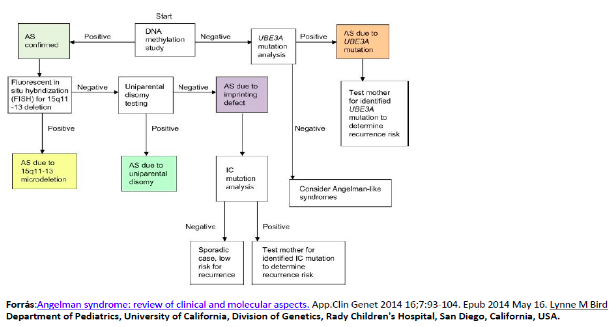
Kivizsgáláskor felmerülő vizsgálatok:
– Angelman szindrómára jellemző EEG eltérések
– koponya MRI vizsgálattal megkésett myelinizáció, fehérállomány csökkenés, fokális fehérállományi eltérések láthatóak (4,5)
– genetikai vizsgálatok:
1. metilációs specifikus PCR
2. Fluorescens spicikus hibridizáció (FISH)
3. Uniparentaralis disomia vizsgálata
4. Imrpinting defektus vizsgálata
Jellemző elektroenkefalográfiai (EEG) eltérések:
– elhúzódó magas amplitúdójú ritmusos 2-3 Hz-es frontális predominanciájú hullámok
– erre interictalis epileptiform hullámok rakódnak
– magas amplitúdójú 4-6 Hz-es aktivitás az occipitalis régióban, tüskékkel, amiknek megjelenését szemcsukás elősegíti (igen jellegzetes, gyakran magában felveti az Angelman szindróma diagnózisát)
– általában a 15q11-13 régió deléciója kifejezettebb EEG eltérésekkel jár (6)
Kutatások során látott eltérések, melyeknek a későbbiekben lehet jelentősége:
-MRI DTI traktográfiával anterposterior és mediolateralis és superoinferior axonalis kapcsolatok is kevéssé fejlettek a normál idegrendszerhez képest (7)
Farmakológiai eltérések:
– 15-ös kromoszóma érintett régiójában a gamma-aminovajsav A receptor alegységének (GABA-A) számos kódoló génje helyezkedik el. Ez magyarázhatja az idegrendszer fokozott ingerlékenységét.
Epilepszia terápiájaként valproát, topiramát, phenytoin, phenobarbital, clonasepam, esetleg adrenocorticotrop hormon (ACTH) használható, általában sikerrel.
Az alvászavaron segíthet a melatonin kezelés (8)
Prognózis:
Az Angelman szindrómás gyermekek és felnőttek jó egészségnek örvendenek és normál élettartam várható, de folyamatos, egész életen át tartó gondozást igényelnek.
A tünetek a korral változnak. Például az alvászavarok felnőttkorra javulnak, serdülőkorban az epilepsziás rohamok is gyakran enyhülnek, ritkulnak.
Elhízás gyakran jelenik meg, mely kiemelt figyelmet igényel.
A családban az ismételt előfordulás esélye a genetikai eltérés függvénye:
– a 15q11-q13 deléciók esetében legtöbb esetben kicsi, 1% alatti.
– Uniparenteralis disomia esetén attól függ, hogy történt-e kiegyensúlyozatlan kromoszóma transzlokáció, vagy nem. (Előbbi esetén egyénileg változó esély, utóbbi esetben 1% alatti ismétlődés.)
– UBE3A génhiba, vagy imprinting defektus esetén a szindróma 50%-ban ismétlődhet. (1)
Az összefoglalót készítette:

Dr. Ambrus Bence
gyermekgyógyász,
osztályos orvos a MRE Bethesda Gyermekkórház Neurológiai Osztályán,
a Down ambulancia vezetője
Forrás:
(1) OMIM #105830
(2) Kishino, T., Lalande, M., Wagstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nature Genet. 15: 70-73, 1997.
(3) Fiumara A., Pittala A., Cocuzza M., Sorge G. (2010). Epilepsy in patients with Angelman syndrome. Ital. J. Pediatr. 36:31 10.1186/1824-7288-36-31
(4) Castro-Gago M, Gomez-Lado C, Eiris-Punal J, Rodriguez-Mugico VM. Abnormal myelination in Angelman syndrome. Eur J Paediatr Neurol. 2010;14:292.
(5) Harting I, Seitz A, Rating D, Sartor K, Zschocke J, Janssen B, Ebinger F, Wolf NI. Abnormal myelination in Angelman syndrome. Eur J Paediatr Neurol. 2009;13:271–276
(6) Laan LA, Vein AA; Angelman syndrome: is there a characteristic EEG? Brain Dev. 2005 Mar;27(2):80-7.
(7) Tiwari VN, Jeong JW, Wilson BJ, Behen ME, Chugani HT, Sundaram SK. 2012. Relationship between aberrant brain connectivity and clinical features in Angelman Syndrome: A new method using tract based spatial statistics of DTI color-coded orientation maps. NeuroImage 59:349–355
(8) http://www.patient.co.uk/doctor/Angelman-Syndrome.htm